


发布时间 : 2018-09-27来源 : 华康基因浏览次数 :
生命的语言
-----DNA和个体化医学
【美】弗兰西斯·柯林斯/著 杨焕明等/译
本书撰自《纽约时报》畅销书作者、世界知名医学专家、遗传学家,弗兰西斯,柯林斯。本书将永远改变你对人体、健康和未来医学的思考方式!
从基因到治疗
研究疾病背后的基因突变是一回事,攻克这些疾病就完全另当别论了。许多人嘲讽说,罕见遗传病的治疗永远不会奏效。来看看镰状细胞贫血症吧!它是最早被发现的第一个隐性遗传病。在祖先来自历史上爆发过疟疾的地方,如地中海地区、非洲和东南亚的人群中,镰状细胞贫血症是很常见的。这种病的基因携带者(即只有一个镰状细胞突变基因的拷贝)反而比正常人更能抵御儿童疟疾的侵袭;而具有两个镰状细胞突变基因的人,会患有严重的血液疾病,他们经常苦痛难熬,寿命也比一般人短得多。
早在50年前,人们就知道镰状细胞贫血症的基因突变发生在编码珠蛋白的一个基因上,但是50年过去了,这一发现对研发新的治疗手段几乎没有任何帮助。那么,我为什么要说遗传医学已经到了一个转折点呢?一方面,医学的进步不是线性的。过去50年的缓慢进步并不意味着未来50年(甚至未来10年)的发展也会同样缓慢。大多数研究人员都相信在未来的10年里,镰状细胞贫血症的治疗将取得长足的进展。他们之所以如此乐观,主要是因为基因治疗的前景,尽管基因治疗这一领域峰回路转,进展缓慢。
要想对镰状细胞贫血症进行基因治疗,我们需要将珠蛋白相关基因的正常拷贝插入到患者的骨髓纲胞中去,并使其在长时间里有效地行使功能。来自其他疾病的研究进展使人们相信,基因治疗最终将有效应用于镰状细胞贫血症。更鼓舞人心的是,基因治疗法并不是治疗手段的唯一突破,还有一些新的理念是开发药物以阻止患者的红细胞镰状化。
那怎么对付囊性纤维化呢?从发现它的病因到现在已经20年了,我们该如何将这一发现应用到治疗上呢?有没有什么值得报道的进展呢?
CFTR基因被发现不久之后,人们对利用基因疗法治愈这种疾病的热情空前高涨。这种想法相当直接:如果我们可以将一个正常基因的拷贝导入一种感染呼吸道的病毒之中,然后让患者“被感冒”一下,这些正常基因就能进入患者体内。我们在发现致病基因一年之后,在实验室里进行了培养皿实验来证明其原理,结果显示这种病毒可以修复呼吸道培养细胞的盐分运输缺陷。然而对于活生生的人来说,将基因进行有效的体内转移仍是一个几乎不可能的任务。障碍主要在以下几个方面:首先,基因的转移效率必须非常高,因为光修复一些呼吸道细胞是于事无补的,因此病毒们得像一个军团一样进行合作,迅速出击并占领大片的领地。其次,把正常基因的拷贝植入细胞后,事情还没完,还必须让它稳定地待在细胞内,并表达足够量的RNA和蛋白质。
最后,我们的成功有赖于如何逃脱免疫系统的监控。因为我们自身的防御功能会迅速消灭“入侵”的病毒,而使我们前功尽弃。
非常不幸,以上提及的问题都是现实的挡路石。为了更好地理解这个问题,找们可以用体育比赛来打个比方,你不妨把你的20000余个基因想象成一支训练有素的队伍,齐心协力来打赢“生命”这场比赛。一个CFTR突变就像一个受了重伤的运动员,不得不将其抬出场外。基因治疗——相当于这场比赛的对策——就是派出一名替补队员上场。但是这名队员必须能够想办法找到自己在场上的合适位置,并且在良好表现的同时保护自己不受伤。假如紧接着他也倒下了并被抬出场外(这种情形就像基因进人细胞后,还没来得及发挥什么功能就壮烈牺牲),那么问题仍然得不到解决。更为严重的是,假如这个倒霉的替补队员不在球队的花名册上,裁判(也就是免疫系统)就会吹停比赛并将他逐出场外。
“路漫漫其修远兮”,基因疗法在过去的25年里一直步履艰难。几年前,成功似乎已唾手可得。当时有那么几个孩子,由于某个基因的缺失患上了一种很罕见的免疫缺陷疾病。以一种人为灭活的病毒为载体为他们补充缺失的基因,患儿们明显得到了治愈。可是仅仅几年后情况就急转直下:这些孩子中有好几个患上了白血病,显然是由当时作为载体的同一病毒无意中激活了白血病相关基因而引起的。我将在第十章里好好聊聊基因治疗。
针对囊性纤维化病的基因治疗仍在继续研发,但是早期那种“一蹴而就”的幻想已经被现在“持久战”的认识所替代,也许还要经过多年的艰苦奋斗。与此同时,囊性纤维化致病基因的发现使得另外一种治疗方法有望成功。让我们再回到刚才那个体育比赛的比喻上,难道我们不能迅速治愈受伤的第一个运动员,然后看着他活蹦乱跳地重新上场比赛吗?这个比喻可能有点过头,但这正是药物治疗的终极目标。
过去,开发药物都是依靠经验。用于制药的化合物基本上来源于自然,此如那些从细菌、真菌或植物中提取出来的物质,并且我们只能检测这些物质中的那么几种。现在,一种新的、更为全面的“药物设计”,正在逐步取代旧的、不够系统的方法。这种新方法会为药物设定一个明确的靶标,然后从成千上万的候选物质中筛选出最有效的那种药物。
就囊性纤维化来说,我们已经精确地知道发生了哪些分子缺失,因此可以通过呼吸道细胞体外培养实验来寻找能够修复盐分运输缺陷的药物。只要这种化合物在动物实验中不表现出毒性,就可以开始临床试验了。有关药物研发的更多信息见附录D。
一种这样的“设计药物”曾在36个囊性纤维化患者身上做过前期临床试验,其结果确实令人振奋。其中一个就是比尔·埃尔德( Bill Elder)。他可不仅是一个年青的囊性纤维化患者,更重要的是斯坦福大学的一名学生,在杰弗里·怀恩( Jeffrey Wine)博士的实验室工作。怀恩博士对囊性纤维化的兴趣,是因为他自己的孩子也被诊断为囊性纤维化患者。相对于其他患者,埃尔德这位“重伤队员”比较“非主流”,其突变是较为少见的G551D(图2.4)。在参加这次试验之前,他的情况不算太糟,尽管他需要服用多种药物,同时每天都必须进行胸部理疗,当然,也免不了隔三岔五的抗生素治疗。
作为本次药物试验的志愿者,埃尔德的代号是VX - 770。他每次服用3粒白色药丸,每天两次,然后还要进行一大堆测试来确认药物是否发挥作用。
这次初步试验的结果让人吃惊。试验对象的汗液含盐量降低到了几乎正常的水平。有一项检测盐分运输的测试是测量鼻腔中的组织,其结果近乎完美。更为引人注目的是,仅仅两周时间,患着肺部的气流情况就大为改善了,而且没有发现这种药物的任何副作用。尽管这只是一次短暂的、前期的试验,但它无疑是药物治疗囊性纤维化的一个里程碑。
目前宣告战胜囊性纤维化还为时过早,研究者们依旧任重而道远。可是这些研究进展给众多患者带来了曙光。
10年前的一次北美囊性纤维化年会上,医护人员、患者及其家属济济一堂。会议上我做了发言,并在报告结束时呼吁大家跟我一起歌唱美好的未来。在场的人们无不热泪盈眶,数千人都站了起来,跟着旋律一起歌唱:
让我们大胆去梦想,
人人都能呼吸顺畅。
我们心如铁来意如钢,
直至病魔成为历史!
这个梦想的实现离我们越来越近了。此外,疾病的新疗法如洪流般不断地从世界各地的实验室里奔涌而出,囊性纤维化上取得的进展是第一个浪潮。而这一切都应该归功于我们解读生命语言的新能力。
从饮食到生命
除了基因治疗和药物治疗,还有一种截然不同的治疗方法就是环境治疗。对于我们而言,饮食是一个重要的环境因素。既然我们明确地知道很多疾病的发病机制,在某些情况下,我们确实可以通过改变饮食来缓解病情。
特雷西·贝克(’rracy Beck)今年35岁,是天体物理学博士,在空间望远镜科学研究所工作,正投身于第二代哈勃望远镜的研制工作。假设贝克早出生10年,就算她现在还活着,多半也会住在精神病院里,智力低下,癫痫频发,头脑发育不全。
贝克出生时,看起来完全正常。但出生后的第一个月里,贝克变得嗜睡,和她姐姐相比更容易困倦,她妈妈为此感到担忧。新生儿体检显示她血液中苯丙氨酸的含量差不多是正常水平的10倍。苯丙氨酸是人体不可或缺的一种必需氨基酸(essential amino acid),所有蛋白质中都有。贝克在遗传上出了一些问魉,导致她的身体无法合成一种酶,即苯丙氨酸脱氢酶,它的功能是催化苯丙氨酸的分解代谢。因此,贝克体内的苯丙氨酸含量高于常人,好东西太多了,变成了坏事情。尽管这种氨基酸是生命活动必需的,但在大脑还在发育的时候,过量的苯丙氨酸只会产生严重的毒性。
诊断的结果令贝克的父母十分震惊。这种病被称为苯丙酮尿症( PKU),患有此病的人终其一生都要不断摄入少量的特定蛋白质,以将苯丙氨酸含量维持在低水平上,同时要保证生长过程所有其他氨基酸的足量摄入。根据业已存在的这种罕见疾病的食疗方法,他们立即着手给贝克制定严格的饮食方案。可以想象,让一个孩子无论是在学校吃午餐或参加生日聚会,还是在朋友家过夜时都保持如此严格的饮食习惯,对她来说是一个多么巨大的挑战。贝克承认她在9岁的时候没有遵守饮食方面的限制,开始偷吃禁忌食物,尤其是奶酪。结果不到几个月,她就跌出了班级尖子生的队伍,甚至有一段时间被安排去了数学补习班。最终她和她的父母意识到如果继续这样下去,可能会毁了她的前途。于是她和父母一起重新制定了她身体承受范围内的食谱,并且努力地让周围的人意识到贝克坚持合理饮食的重要性。时至今日,她仍旧保持着那套不寻常的饮食习惯。当出席社交场合时,其他人都在大快朵颐,尽情享受那些高蛋白的美味佳肴。可她却简单地告诉朋友“我有医学上的问题,不能吃这些蛋白质”。
这是一种极端的限食方案,远远超出一般人控制胆固醇摄入的忍受程度。例如贝克要避免饮用任何含有天冬甜素的“健恰”碳酸饮料,因为这种人工合成的甜味剂会在体内转化成苯丙氨酸,这会给PKU患者造成灾难性的后果。尽管身体受到严格的限制,贝克仍然取得了巨大的成功,她是第一批获得博士学位的PKU患者之一,是PKU年轻患者学习的楷模。对她而言,一个重大挑战是说服保险公司为她特殊的食谱买单,这份食谱每个月需要花费大约1300美元。其实傻子都能看出来,保险公司应该毫不犹豫地为这种被科学证实的、高效的,如不治疗即后果严重的疾病治疗方案买单,但是这种令人信服的逻辑并不总能说服美国目前的那套运作不良的系统。
PKU是一种隐性遗传病,这样说来她的父母肯定双双都是携带者。贝克的两个弟弟在出生后没几天就被诊断患有PKU。这正好证明了遗传风险计算的要点。如果父母均为隐性致病基因的携带者的话,生下的每个孩子患病的风险是1/4(图2.2)。而风险是没有记忆的,因此一个拥有4个子女的这样的家庭里就应该有0—4个患儿。在贝克家,4个孩子中却有3个患儿(只有一个姐姐免遭厄运)。她的两个弟弟也很好地控制了各自的饮食,目前都已经大学毕业,正在从事通讯方面的工作。
综上所述,在所有那些100%遗传的,但又能通过环境控制进行100%预防的疾病中,苯丙酮尿症的食疗是目前最具有说服力的例子。
最近,一个五岁半的小男孩为遗传疾病的药物治疗的巨大进展提供了第二个绝好的案例。他叫布莱克·埃雷胡斯(Blake Al-thaus),从他出生起就不停地有人称赞他那修长优美的手指,甚至据此想象出一个未来的天才钢琴家。然而,当他妈妈注意到他的脊椎可能存在一个不正常的夸曲时,她就开始为他担忧了。紧接着一位眼科医生发现了另一个问题:眼球晶状体错位。但是最严重的问题是在一次心脏超声波检查中发现埃雷胡斯的主动脉,即直接从心脏出来的人体内最大动脉的第一段出现了膨大,并且在将来有可能突然破裂,进而导致猝死的风险极高。
埃雷胡斯的父母被告知,他们的孩子患有被称为马方综合征(Marfan syndrome)的严重疾病,而且是特别严重的那种类型。鉴于孩子的大动脉正在快速膨大,一位医生预测说他很可能活不过两岁。万念俱灰的父母在网上四处搜索,终于联系上了约翰-霍普金斯大学的迪茨( Hal Dietz)博士,一位在马方综合征领域享有全球盛誉的专家。迪茨博士告诉他们,那位医生的预测实在是过于悲观了,但同时也警告说埃雷胡斯需要深度监护并且很可能需要对正在膨大的大动脉立即施行手术。
事情就在此时峰回路转。从一开始的细胞培养实验到后来的马方综合征小鼠模型,迪茨博士已经对这种病进行了多年的研究。最终,他等到了一种可能延缓甚至阻止大动脉受损的药物。更棒的是,这种名为氯沙坦( losartan)的药物用于治疗高血压已经有十多年的历史了,并且儿童可以安全服用。
就这样,只有18个月大的埃雷胡斯与氯沙坦结缘了。他的父母揪着心等待着结果。直到此时,超声波检测的结果显示他的大动脉仍在膨大,危险仍在继续。几个月过后,这种膨大停止了。在接下来的4年中,埃雷胡斯的大动脉状况不断地改善。现在他已经五岁半了,他的大动脉基本上跟同龄的正常人一样。
马方综合征是由原纤维蛋白(fibrillin)基因的一个突变引起的。原纤维蛋白是构成结缔组织的一种不可或缺的物质。结缔组织包括大动脉、脊椎,以及将晶状体固定在眼球正确位置的那些纤维。这一突变在20年前首次被发现的,大多数研究人员都认为它是极难用药物治愈的,因为修复一个结构性蛋白质要比补偿一个催化某个代谢途径的酶类蛋白质困难得多。这就像是一栋用劣质砖头建造的砖房,你要是想在里面住得安心,就得一块一块地把这些劣质砖头找出来,然后一块一块地替换掉。不过迪茨博士和他的团队向这种保守观点发起了挑战,最终他们证实原纤维蛋白还有另一项重要功能:它与一种名叫TGF-beta(转化生长因子-β)的蛋白质相结合。在马方综合征中,原纤维蛋白出了问题,于是TGF-beta蛋白在体内的含量开始异常地增加。根据研究人员提出的假说,这种内源的“过度剂量”可能会导致大动脉膨大。这也是尝试氯沙坦的原因,因为这种降血压药物能额外地作为TGF-beta的拮抗剂来使用。在早期进行像埃雷胡斯这样的重症患者的试验中,结果好得出入意料。
目前正在进行一项大规模的临床试验,这次试验的目的是观察氯沙坦是否能缓解不像埃雷胡斯那样严重的成人患者的病情。患有马方综合征并因为大动脉破裂而猝死的知名人士有:排球明星海曼(Flor Hyman),以及百老汇名剧《房租》的作者乔纳森·拉尔森(Jonathan Larson)。随着氯沙坦的使用,许多类似的悲剧有望避免发生。
从“要你做”到“我要做”
(谁想对你进行筛查?原因是什么?)
很多疾病,像苯丙酮尿症( PKU)和囊性纤维化(CF),现在可以根据特定的生化或DNA检查的结果,对发病的概率作出非常准确的预测,并已经有办法进行医疗干预。有时候,是检查某一个人,以判断被测试者是否患有需要治疗的遗传性疾病;有时候,是检查准父母,来判断他们是否携带了那些虽然不会影响他们的健康,但有可能咸胁到下一代的基因突变a在医学发展的新时代,我们把“遗传筛查”(genetic screerung)这个术语,用来描述大规模的人群“测试”,它不需要知道你的家族病史或既往病史。遗传澳6试(genetic testing)则更有针对性,应用于当你感到有高危的异常情况出现时。
新生儿筛查
贝克,一个患有苯丙酮尿症的天体物理学博士,代表了新生儿筛查技术的一个十分成功的案例。早在20世纪60年代,美国的大部分州就已经开始了苯丙酮尿症的筛查。随着时间的推移,越来越多的疾病已经被列入筛查清单。人们关注的重点是那些筛查技术成熟的疾病,以及那些早期诊断肯定会带来好处的疾病。很多时候,这些诊断结果可以指导药物治疗,有的时候可以辅助设计特殊的食谱,还能为外科手术提供建议,或发挥其他作用。
美国“出生缺陷基金会”(March of Dimes,又称“一角钱运动”)现在推荐对新生儿做29种疾病的筛查。每年有大约4000个新生儿被发现患有其中的一种疾病。美国所有的州都会为新生儿筛查苯丙酮尿症、甲状腺功能低下症、半乳糖血症,以及镰状细胞贫血症。
筛查是非常有必要的。甲状腺功能低下症不只是由单一的遗传因素引起的,因此早期检测至关重要。如果你的孩子患有甲状腺功能低下症,你必须立即采取甲状腺激素替代疗法来促进大脑的正常发育。半乳糖血症是由一种阻断半乳糖代谢的突变引起的。奶中含有的半乳糖,需要转化为葡萄糖才能被吸收。这样的情况可以通过限制饮会来治疗。镰状细胞贫血症(前文已提及)在美国非洲裔的婴儿中的发病率为1/400,早期诊断能警示及时的治疗。这些治疗包括为患儿使用疫苗和青霉素来降低细菌感染的危险,因为镰状细胞贫血症患儿特别容易被感染。
很多州也为婴儿进行囊性纤维化的筛查。大量证据表明,早期诊断能带来更好的药物治疗和更合理的营养状况。
你能在这个网站上找到“出生缺陷基金会”所推荐的全部婴儿筛查项目表:http: //www. marchofdimes.com/professionals/14332—15455. asp。表中非常重要的一项——听力缺陷,每1000个新生儿中,大约有两三个婴儿会患这种病。先天性听力缺陷可能由多种突变引起,也可能由非遗传原因引起。如果没有早期的筛查,这种疾病的患儿很可能在出生后的很多个月里都没有被诊断出来。而在这段时间里,患儿的听力和语言能力可能会受到几乎不可逆转的损害。
随着医学研究的进步,这份疾病筛查清单将会变得越来越长。现在的新生儿筛查只需要从婴儿的脚后跟采几滴血,用滤纸吸附后送到中心实验室进行分析。一些州已经开始采用更加先进的技术来检测涉及氨基酸、有机酸、血糖等指标的多种疾病,甚至超过了“出生缺陷基金会”推荐的筛查范围。
有时候,这样做也会产生一个现实问题。在某些新生婴儿身上,可能出现一些以前没有记载的异常状况。这些状况中的一部分是无害的,然而另一些却会导致智力低下,甚至是致命的。如何处理这些意义不明的代谢疾病的挑战,让医护人员伤透了脑筋,也让父母们提心吊胆。尽管如此,但有一点毋庸置疑,新生儿筛查能够显著提高那些可治疗的遗传病昀早期诊断。
几乎可以肯定,新生儿筛查势必发展成一个更为广泛和全面的检查方式。随着全基因组测序成本的不断下降,在未来5—7年里,很可能会降到1000美元以内。对于是否该在新生儿出生时就获得全基因组信息的争论愈发引人注目。这种检查或许会引起我们当中某些人的焦虑。1997年上映的电影《加蒂卡》①有这样一幕场景:在一间高科技的产房里,电影中的男主角在这里出生了。基因组分析已即刻完成,未来的预测随之而来:非常精确而又十分恐怖。这绝对不会是我们的未来!基因并不能决定我们的命运,尤其是对于心脏病、糖尿病、癌症这样的常见疾病来说。但是,一个温和版本的《加蒂卡》或许会在不久的将来问世。
我在本书的后面章节会用更多的文字来讨论“遗传隐私”和对于未来风险的“不知之权”。毕竟,一旦某个人的DNA序列被确定了,它的主人就失去了要说“不,谢谢”的机会;另一方面,当我们对遗传风险因子的有效干预知道得越来越多,并且认可在生命早期进行干预可以带来明显好处时,在出生时了解遗传信息将变得越来越引人关注。一个可能的折衷方案是:想出一个办法对那些不是非马上知道不可的信息进行保密,等孩子长到18岁之后让他们自己决定想知道什么。
当想起一个与《加蒂卡》类似的未来时,很多人都会不由自主地产生畏避之想。但是来看看肥胖症吧。肥胖症高度遗传,据目前的估计,一个成年人体重的60%—70%是由基因决定的。已经发现好几个这样的基因。如果你生下~个有肥胖症高度遗传风险的孩子,你最好从婴儿时期就开始改善f也/她的饮食;而不是过五六年后才发现他/她已经超重并养成了暴饮暴食的不良习惯。
①《加蒂卡》(CATTACA),也译为《千钧一发》、<变种异煞》,是一部科幻片。它说的是“不久的将来”,经基因选择的孩子才是人类的精英,而自然出生的则被视为“上帝的孩子”或“病人”。男主角文森特正是这样的“病人”,但他想参加加蒂卡太空企业的太空计划,于是假冒精英的基因身份,并躲过重重追查,最终实现了飞上太空的梦想。
携带者筛查
隐性遗传病的携带者通常是完全正常的,但是两个携带者的孩子有25%的概率会患病(图2.2)。推动携带者筛查的第一个热潮是黑蒙性家族痴呆症(Tay-Sachs disease)。患者主要多见于具东欧犹太人(Ashkennazi,德系犹太人)遗传背景的人群,但不限于此。患有这种病的婴儿在生命的头6个月里表现正常。但是在那之后,一种他们不能代谢的储存物质在大脑中堆积,病情开始持续恶化,包括失明,耳聋和瘫痪,患儿一般在四五岁时夭折。这种病是由于氨基己糖苷酶A的缺失引起的。在1970年,人们发明了一种酶检测技术。根据检测结果,差不多每30个东欧犹太人中就有一个是这种疾病的隐性携带者。
20世纪70年代,在经过广泛的社区咨询后,犹太人社区开展了携带者筛查,此事引起了浓厚的兴趣。夫妻双双被发现为黑蒙性家族痴呆症基因携带者的,一般都希望知道结果,以便做出生育决定,从而避免患上这种可怕疾病的孩子出生。携带者夫妻通常有以下几个选择:要么收养孩子;要么采用非携带者捐献的精子或卵子进行人工授精;要么考虑他们的意愿,进行产前诊断并结合可能的终止妊娠。
携带者筛查在犹太人社区十分普及,患有黑蒙性家族痴呆症的新生儿几乎绝述。具有讽刺意义的是,这一疾病现在多见于其他族群的儿童。尽管他们的突变率虽然远远低于犹太人,但没有开展黑蒙性家族痴呆症的携带者筛查。
70年代还制定了镰状细胞贫血症的携带者碲查方案,由于非裔美国人群中每10个人就有1个是携带者。但是这一次,结果却不是那么成功,尽管用心良苦,还得到了非裔美国人团体领导人的支持。镰状细胞贫血症的患者和携带者之间的区别非常含糊,统称为镰状细胞性状。镰状细胞性状基本上不会对个人健康产生影响,除非是在极端的环境下,譬如在高空飞行的没有加压舱的飞机中。但个中原因现在也无法解释清楚。更糟糕的是,尽管早在70年代,携带者夫妻就能通过简单的筛查检测就能检出,但是仍然没有有效的产前检查。因此携带者夫妇的选择会比黑蒙性家族痴呆症少得多。还有一个情况:提供筛查的常常是白人,而接受筛查的却总是黑人。这唤起了人们关于优生学运动的梦魇。大部分携带者筛查计划最终都关门了之。
囊性纤维化遗传基因的发现带来了一个机会,可以向夫妇提供囊性纤维化患儿出生风险的信息。但这种事情并不是毫无争议的。毕竟囊性纤维化患者的存活率在逐年稳步上升(图2.5),所以它与黑蒙性家族痴呆症还是有天壤之别。尽管如此,90年代的调查研究显示,很多夫妇还是有兴趣得知这些信息。
而这里仍然存在一个很大的问题是,我们的医疗系统没有起到鼓励个人或夫妇来做孕前筛查的作用。目前,囊性纤维化的携带者筛查几乎总是发生在第一次找产科医师做产前检杏时,但此时怀孕已成事实。按照我们大多数人的观点,采用黑蒙性家族痴呆症的筛查策略,即在夫妻计划怀孕之前就进行携带者筛查更为可取,因为筛查结果能为他们预留更多的选择。
假如我是一个准备组建家庭的年轻人,我会对自己进行筛查,同时鼓励我的太太也这么做——不只是筛查囊性纤维化,还有一长串隐性遗传病呢!目前,美国地区每1000个孕妇中就有1人会有遗传病的麻烦,而这本来是可以据携带者筛查而预测出来的。你会惊奇地发现这些诊断往往带来巨大的震惊,因为一个隐性基因可以代代相传而不露任何迹象。但是,我们目前这种模式,将携带者筛查推迟到怀孕之后,才让夫妻们来做这样无奈的抉择,这样剥夺了他们选择更好的孕前替代方案的权利。
对于携带者筛查还有很多东西可谈。究竟筛查有多可靠?你也许还记得,囊性纤维化几乎总是由CFfR基因的突变引起的吧,这种突变有1000多种不同的“错拼”方式。为了筛查出尽可能多的携带者,同时让价格也不至于高得离谱,现在的大部分筛查只检查CFIR的23种最常见的突变,其结果是能筛查出大约90010的携带者。这就意味着,即使夫妻中的一方的筛查结果为阴性,他们还是有可能生下一个囊性纤维化患儿。
没有得到被测试者充分的知情同意,携带者筛查是绝对不允许进行的,对已怀孕的携带者筛查时尤其要注意这一点。不管如何,对于那些沉浸在幸福中的准父母来说,得知腹中的宝宝有可能患上囊性纤维化或其他隐性疾病会让他们面临艰难的选择。如果父母在孩子出生前并没有兴趣知道有关孩子患各种疾病的概率,或如果说他们根本不会因为任何情况而终止怀孕,那么他们完全有理由拒绝这种测试。
然而,携带者筛查并不一定意味着必须终止有问题的妊娠,让人们认识这一点也是很重要的。有这样一些准父母,他们也希望得到这些信息,其目酌是为宝宝准备特殊的健康护理。
在不远的将来,如果在怀孕之前仍然不能得到所有夫妻的全基因组序列,要不要考虑其他的携带者筛查技术呢?一项正在考虑的筛查是针对脊髓性肌萎缩( SMA)。这种隐性遗传病的患儿在刚出生时表现正常,但就在随后的几个月内,他们逐渐丧失肌肉张力,进而发生完全的弛缓性麻痹,最终在两岁时夭折。大约每40个人中有1个是SMA的携带者,这就意味着在1600例妊娠中就有1例是高风险的,而其中有1/4将成为患儿。就像所有的隐性遗传病那样,携带者并没有症状,且通常也没有家族史。鉴于这种疾病的严重性,筛查SMA的重要性不亚于黑蒙性家族痴呆症。不幸的是,这一筛查十分复杂。SMA是因为这一重复基因的全部拷贝的缺失引起的,要检出突变,只有对DNA大片段进行细致的定量分析。现有的检测能找出94%的携带者,但一次需要花费数百美元。
另外一种正在讨论的是脆性X综合征携带者的筛查。其命名是因为该病的男性患者的X染色体通常会有一个可见的脆性位点。将实验室培养的细胞用特殊的化学试剂处理,在显微镜下可以观测到这一脆性位点。一直到1990年,这种复杂且棘手的分析是唯一可用的检测方法。就在那时,这种疾病在分子基础被发现:患者X染色体上的一个特定基因失活了。其原因是该基因的“上游”有一连串的“CGG”的“串联重复” (tandem duplica-tion)。正常个体的CGG重复不超过45个;如果超过200个,这个基因就会被有效关闭。由于该基因位于X染色体,而女性有两条而男性只有一条X染色体,所以男性的患病率要比女性高得多。
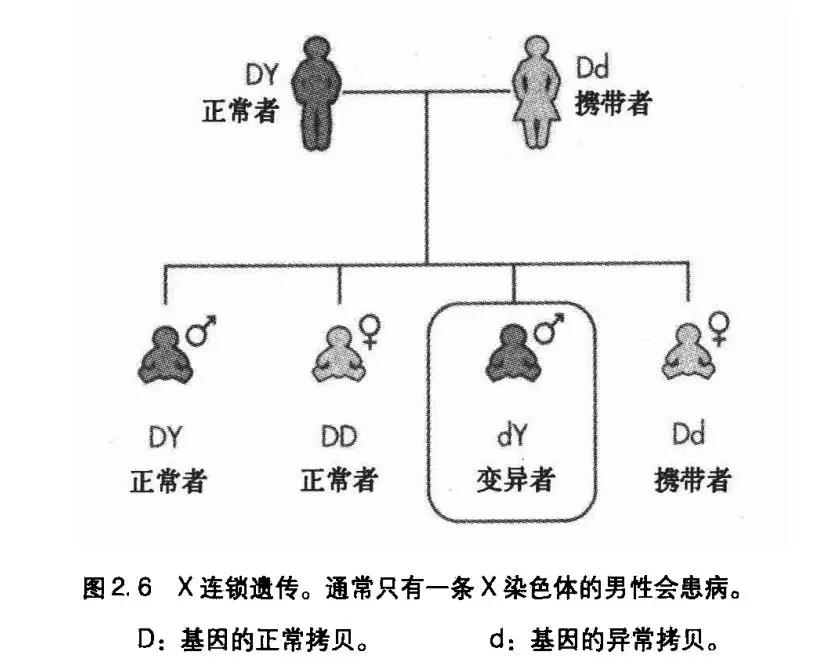
图2.6显示了一种典型的X迮锁遗传。女性也许是某-X连锁的隐性遗传病的携带者,但她们一般不会得病,因为她们还有一条正常的X染色体。女性携带者的儿子会有50%的概率患病。从来没有发现“男传男”的例子,因为父亲传给儿子的只有Y染色体,而不是X染色体。
脆性X综合征是导致智力低下的第二大病因,仅次于唐氏综合征。大约4000名男性中就会有1位患者。该病在所有族群中都会发生,通常没有家族史。此外,这种X连锁的隐性遗传病会有一些特殊:约1/3的女性携带者会出现轻度的学习障碍甚至轻微的智力低下。
考虑到这种疾病的重要性、携带者的出现频率以及DNA检测的可行性(尽管技术上并不简单),为所有女性进行脆性X携带者筛查的呼声愈来愈高。但现在关于如何推行这一计划尚未形成统一意见。
对携带者筛查适当性的辩论在未来几年或许会发生质的改变,这是因为越来越多的个人将会拥有他们完整基因组的全部DNA序列,这将能揭示他们的全部携带者风险,并为夫妻们提供一个在开始怀孕前知道这些风险的机会。很可能几十年后,当人们回顾我们现在的情况时,会对我们只筛查那么几种疾病感到不可思议。他们还会像我现在一样感到困惑和失望,因为我们的医疗系统不能对夫妻进行孕前携带者筛查,这把许多夫妻推向了不必要的两难境地。
Copyright © 2016 中关村华康基因研究院培训 . All Rights Reserved京ICP备18042576号-2


