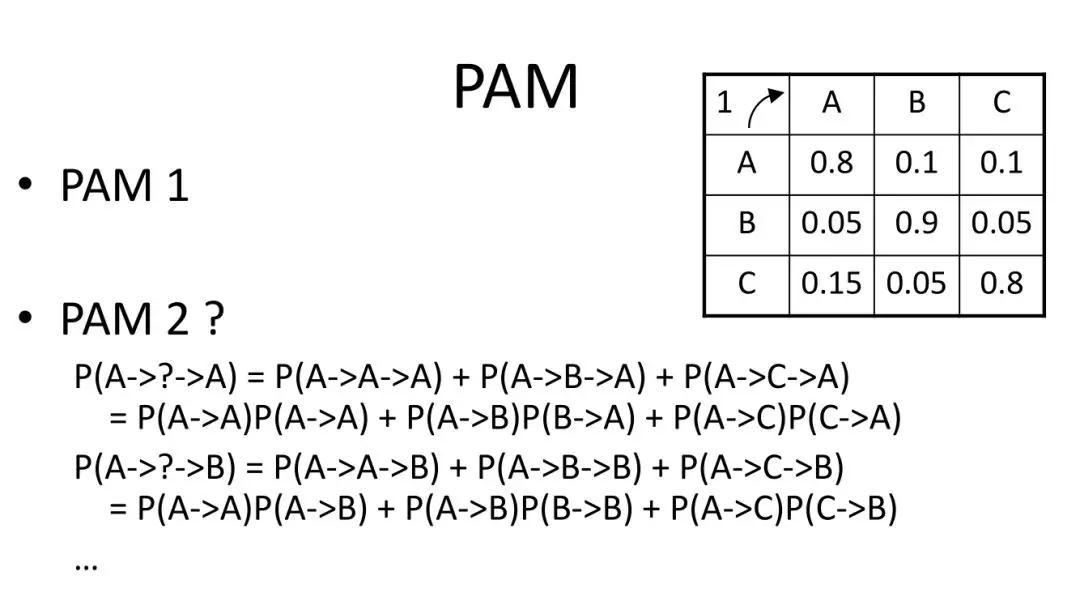
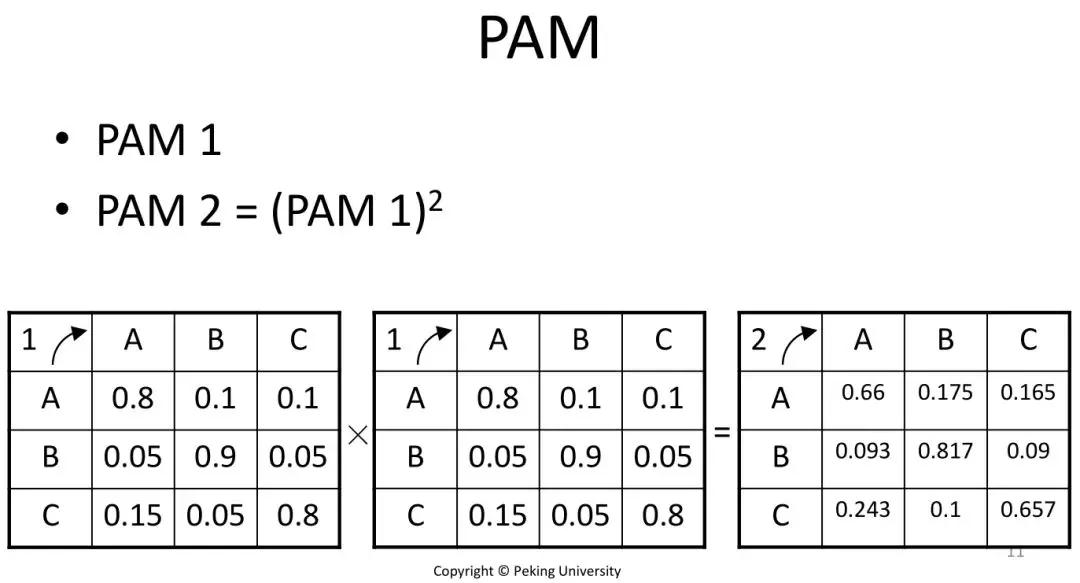
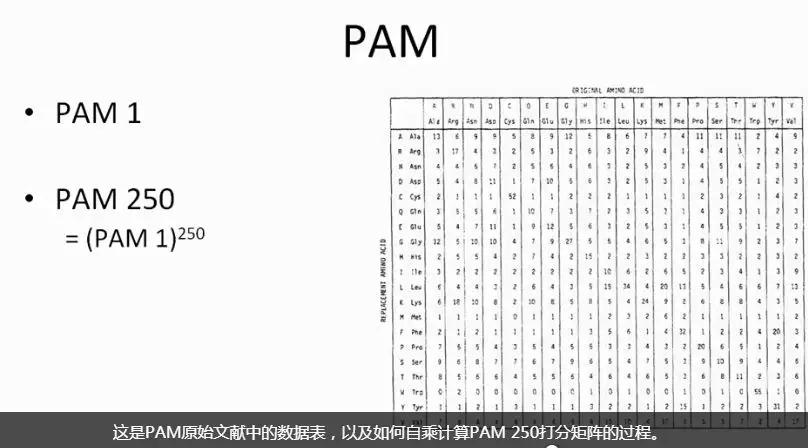
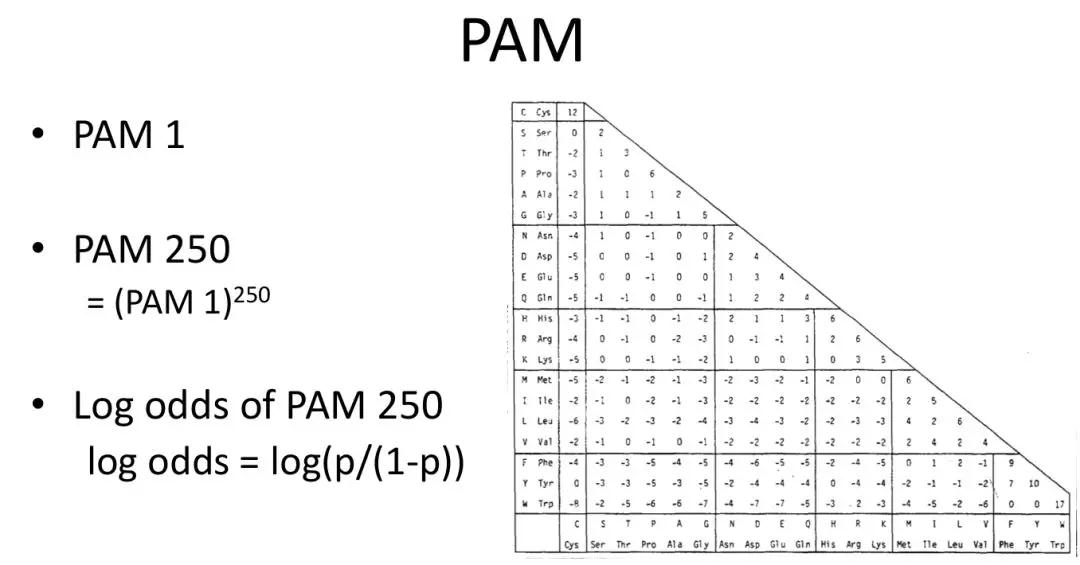
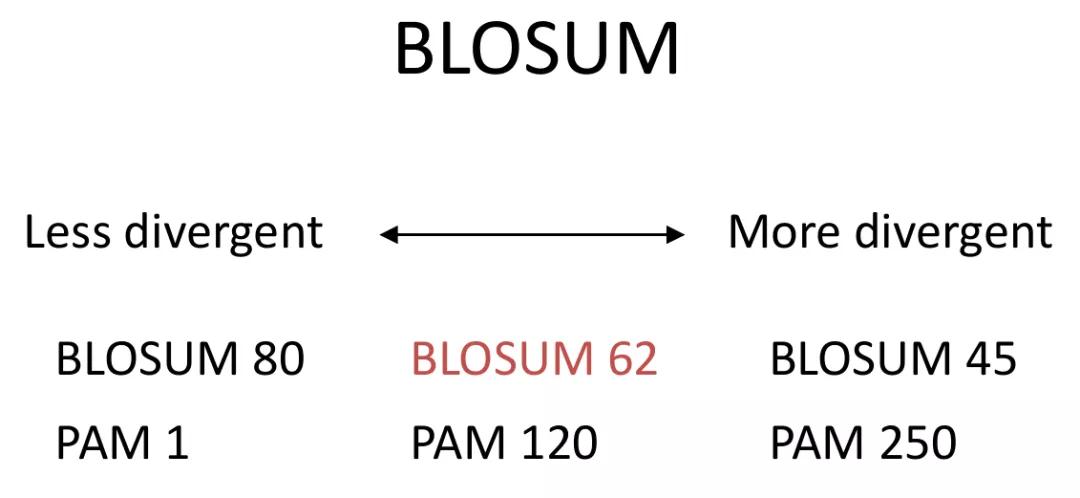
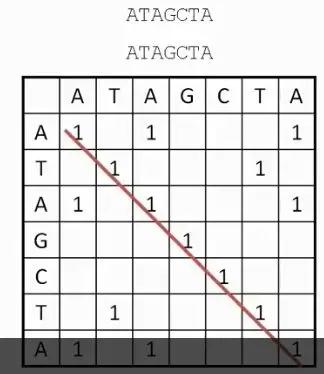
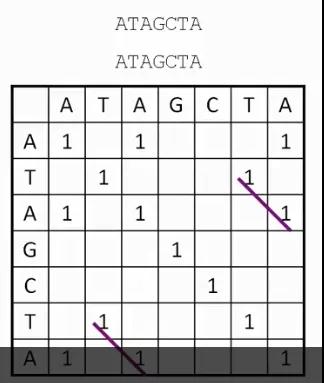
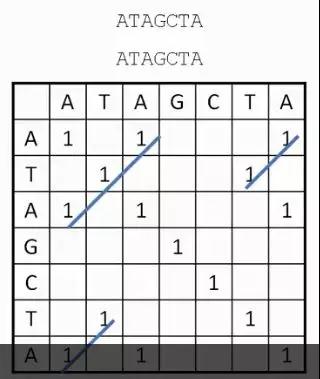
发布时间 : 2019-01-17来源 : 中关村华康基因研究院 浏览次数 :
上一篇 : 121种罕见病诠释(七)
下一篇 : 染色体微阵列分析技术(CMA)
法律声明|公司概况
Copyright © 2016 中关村华康基因研究院培训 . All Rights Reserved京ICP备18042576号-2