


发布时间 : 2019-01-22来源 : 中关村华康基因研究院 浏览次数 :
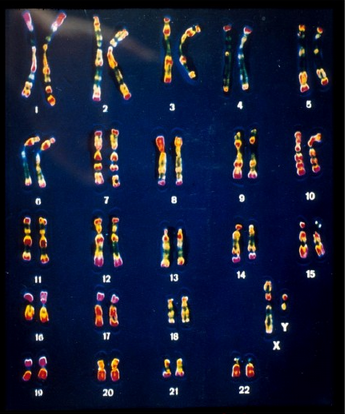
也称为性腺发育不全症。患者外貌象女性,第二性征发育不良,卵巢发育不全,原发性闭经,无生育能力。身体较矮,身高在1.2—1.4之间,肘外翻,常见盾状胸,35%伴有先天性心脏病智力低下或正常。
女性中的发病率约为4/1000。它们的体细胞染色体数是45,比正常女性少了一条X染色体,性染色体组成是X0,一般记作45,X0。
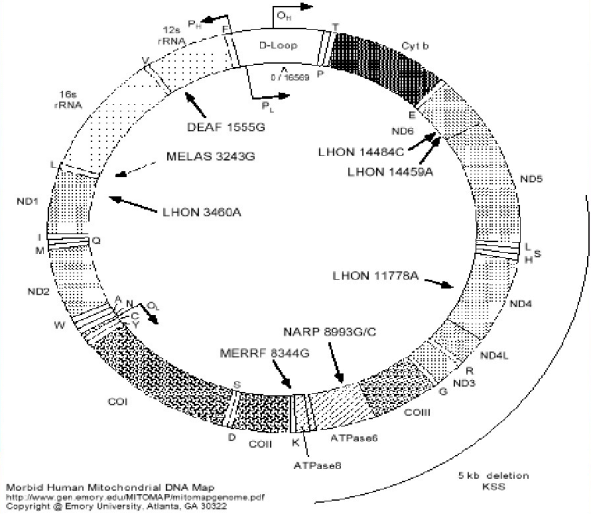
62、Leber遗传性视神经病变(Leber herditary optic neurpathy, LHON)
是一种以母系遗传为特征的线粒体遗传病。
分别突变于:1178位点、14484位点、3460位点。


临床症状:
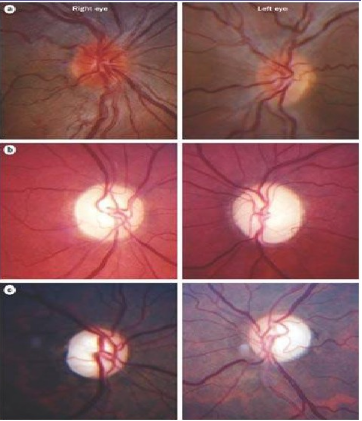
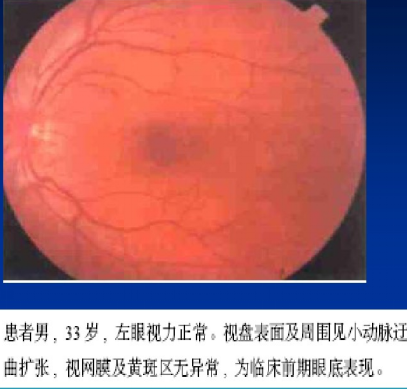
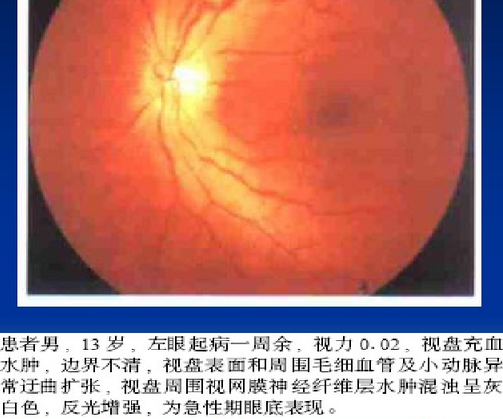
眼底有特征性的三联征:
环绕视盘的微血管扩张弯曲
视盘及其周围神经纤维层水肿
荧光素眼底血管造影(FFA)无血管荧光素渗漏(如右图)
63、长链3-羟酰基辅酶A脱氢酶缺乏症
脂肪酸代谢氧化病:短链、中链、极长链酰基辅酶A脱氢酶缺乏症。
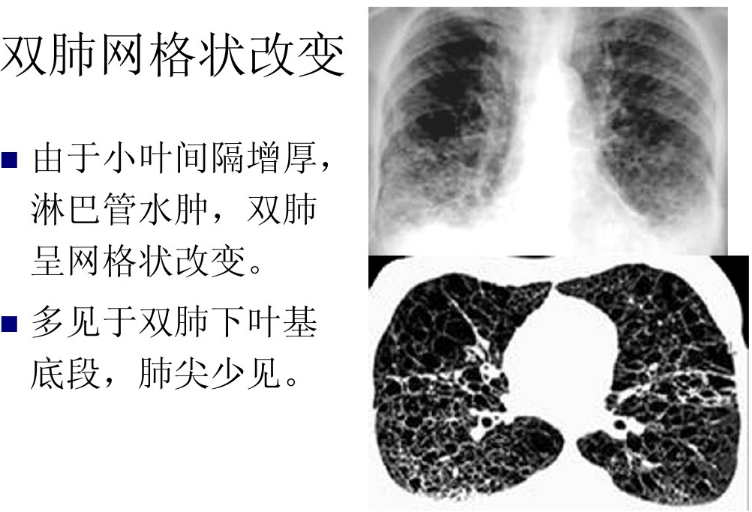
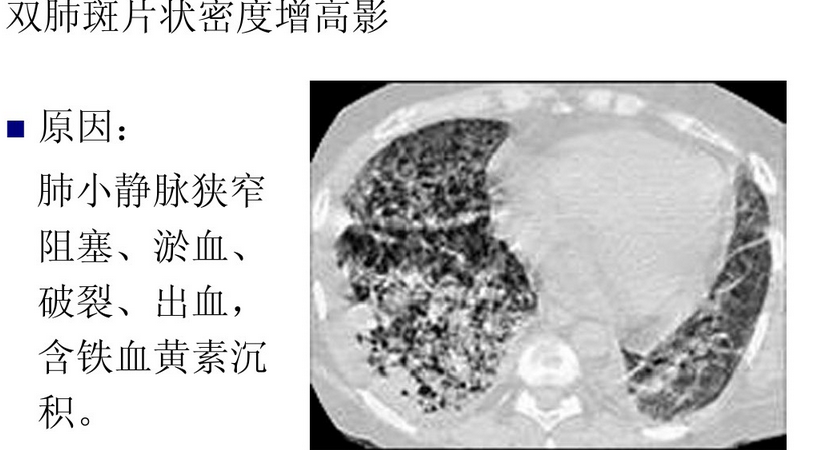
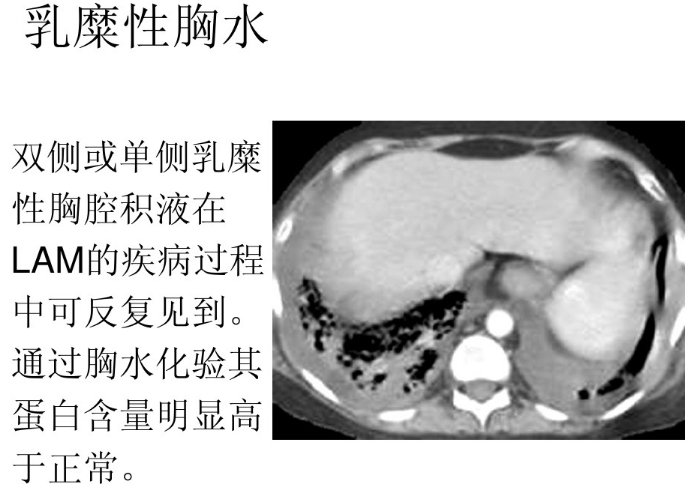
65、赖氨酸尿性蛋白耐受不良( lysinuric protein intolerance , LPI)
为罕见的常染色体隐性遗传病,使赖氨酸转运发生障碍。1965年在芬兰人群中首次报道,是尿素循环障碍的一个亚型,本病在芬兰地区有流行趋势,发病率为1/76000,在意大利及日本南部也有丛集性发病的报道但目前国内报道少见。
临床表现:
婴儿拒食
生长障碍
肌张力低下
反复发作的呕吐和腹泻
66、溶酶体酸性脂肪酶缺乏症
(Lysosomal Acid Lipase Deficiency)
溶酶体酸性脂肪酶缺乏症:是一种内分泌与代谢疾病。常染色体隐性遗传。突变位点位于10号染色体长臂端(10q23.31)LIPA 基因4号外显子杂合突变 c.253 C>A. c.294 C>G
临床表现:主要变现为胆固醇酯和甘油三酯的积累,并且主要是在巨噬细胞中。患者缺乏溶酶体酸性脂肪酶,血清转移酶升高,肝肿大,肝脂质堆积和血脂异常.体重增加,腹部凸起,肝纤维化,营养不良,肠道吸收不良。有2种不同的表型:酸性脂酶缺乏症(WD)和胆甾醇酯贮积疾病(CESD)。
一种常染色体隐性遗传病,是由于支链氨基酸α-酮酸脱氢酶复合体基因缺陷,使支链氨基酸BCAAs(亮氨酸、异亮氨酸和缬氨酸)转氨基反应后形成的相应支链α-酮酸BCKAs不能氧化脱羧,组织中BCAAs和BCKAs异常增高,体液排出带有枫糖浆香甜味的BCKAs而得名。
疾病突变位点
遗传缺陷在线粒体中存在的BCKD多酶复合体中的E1、E2和E33个亚基的基因有突变。
E1α亚基基因的外显子7有C→T转变,结果E1α亚基有精氨酸R242X的无义突变,即E1α在242位精氨酸位点即停止编码。
E1β亚基也可发生突变。其中一个来自母亲的突变基因,在核苷酸编码序列的第526位有A→T突变,另一突变的等位基因来自父亲的突变基因。在第970位有C→T突变而形成终止密码子。
E2基因突变有:插入突变,即在外显子5′端插入17bp;外显子2缺失2个bp,外显子8的5′端的(donor)位缺失1个bp,结果使外显子8整个缺失;外显子8最后1个核苷酸有G→A突变。其他点突变还有外显子7有T→G突变,外显子6有G→T突变,
除内含子缺失突变外,还可有其他突变:
在内含子8有A→G单个碱基取代而创造了5′端有1个新的剪接位点;
2.在外显子11的第1463位核苷酸有G→T转变而形成终止密码子。
3.复合性杂合子,在外显子A的第309位有G→A转变,同时在外显子9的1165位有C→G转变。
E2基因突变有:插入突变,即在外显子5′端插入17bp;外显子2缺失2个bp,外显子8的5′端的(donor)位缺失1个bp,结果使外显子8整个缺失;外显子8最后1个核苷酸有G→A突变。其他点突变还有外显子7有T→G突变,外显子6有G→T突变,
除内含子缺失突变外,还可有其他突变:
在内含子8有A→G单个碱基取代而创造了5′端有1个新的剪接位点;
2.在外显子11的第1463位核苷酸有G→T转变而形成终止密码子。
3.复合性杂合子,在外显子A的第309位有G→A转变,同时在外显子9的1165位有C→G转变。
E3是同源二聚体黄色素蛋白(flavoprotein),为所有α-酮酸脱氢酶复合体成员所共有,它是一种特异性激酶,E3基因座定位在7q,也可突变,但比E1和E2少见,E3基因突变除支链α酮酸脱氢酶缺乏外还有丙酮酸脱氢酶和α-酮戊二酸脱氢酶功能受损。
临床表现:
1.经典型枫糖尿症
患儿在出生时状况良好,一般从生后几天或1-2个月内发现喂养困难,啼哭声弱,不能吸乳和反应迟滞,以后即逐渐消瘦,智能低下,同时呼吸变浅,间断出现发绀现象。小儿囟门常膨出而紧张,还有时出现眼震,眼肌瘫痪、睑下垂、瞳孔散大及对光无反应等。
2.间歇性枫糖尿症
早期发育正常,反应也不迟钝,大约从生后10个月到2岁间歇性出现厌食、呕吐、表情淡漠、步态不稳、共济失调、嗜睡和行为改变等。尿中有特异的气味。
3.轻型枫糖尿症
表现为精神发育迟滞,但无其他典型神经症状和体征,也没有间歇发作的特点。
4.对VB1反应型枫糖尿症
仅有轻度智能发育迟滞,也无典型的或间歇神经损害症状。仅有血中支链酮酸的含量比正常儿稍高。本型患儿对维生素B1的疗效好,血中的生化异常可有显著的好转。
5. (E3)缺乏型枫糖尿症
极为罕见,临床表现类似中间型,患儿在出生数月内通常不出现症状,随着病程进展,逐渐出现进行性的神经系统症状,如肌张力减低、运动障碍、发育迟滞等。
疾病突变位点:人类第15号染色体上有一个叫做FBN1的基因,编码微纤维蛋白1。它可以被看成是把身体细胞黏合在一起的胶水。这个基因发生突变后,“马凡氏综合症”便产生了。
临床上疾病表现
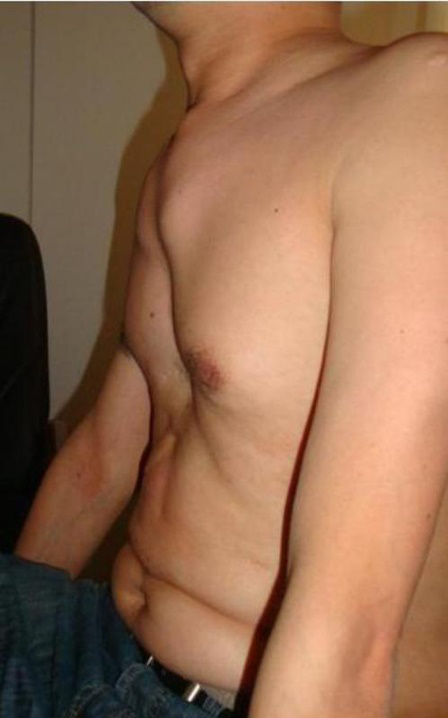
1.骨骼改变:四肢奇长且细,尤以指(趾)为著,肌肉张力降低,关节活动增加,可有超常的运动范围,但脱位罕见,头长,额部圆凸,胸骨畸形多由肋骨过长所致漏斗胸或鸡胸更常见,肩胛隆起呈翼状。全身性结缔组织异常可累及关节囊、韧带、肌腱、肌膜,可导致关节反复脱位、扁平足或高弓足,腭弓高,牙齿不整齐。
2.皮肤改变:最常见的皮肤表现为皮纹增宽或有萎缩性皮纹这些皮肤异常表现可见于身体的许多部位,尤以胸部、肩部三角肌区和大腿部为显著。
3.心血管异常:30%~40%的患者有心血管系统并发症,最常见的心血管异常为主动脉特发性扩张、主动脉夹层动脉瘤和二尖瓣异常
眼部改变:最特征性表现是晶体脱位或半脱位,约3/4的患者为双侧性。
5.神经系统病变:表现为蛛网膜下腔出血和颈内动脉瘤所致的压迫症状动脉瘤引起的癫痫大发作。
症状表现

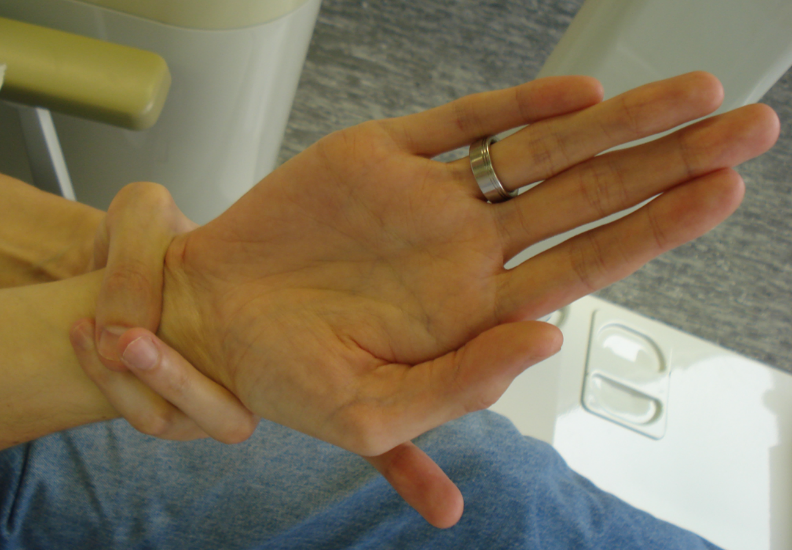
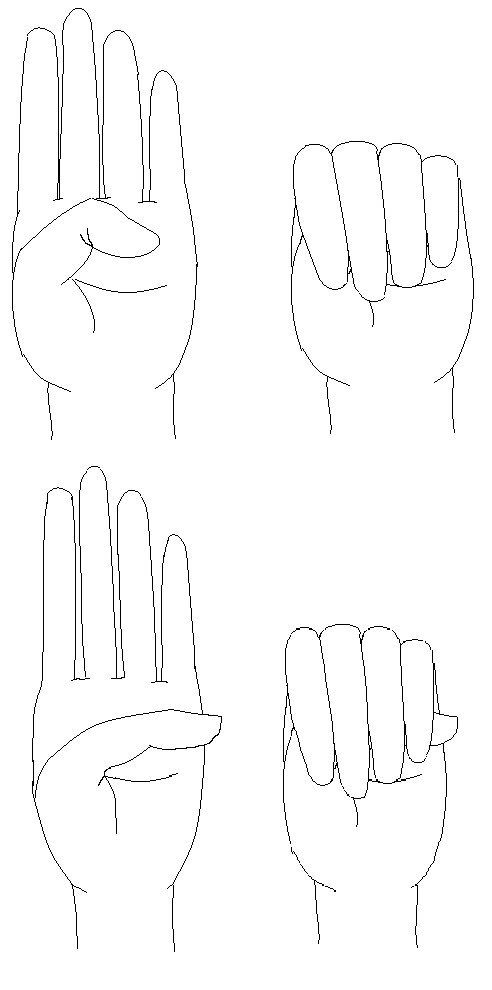
疾病突变位点:本病的遗传学基础是在胚胎形成过程中的鸟嘌呤核苷酸结合蛋白( G蛋白) α亚基( Gsα)基因的突变。常见的突变是位于20 号染色体长臂的编码Gsα亚基基因8号外显子的Arg 201 His或Arg 201Cys错义点突变。
临床上疾病表现
①一个或多个内分泌腺增生或腺瘤引起的自主性功能亢进。
②多发性骨纤维异样增殖。
③边缘不规则的皮肤咖啡色素斑。


是一种常染色体隐性遗传病, 主要表现为线粒体脂肪酸的β氧化异常,并出现一系列相应代谢指标异常。急性发作时,常表现为低酮性低血糖、呕吐,其他症状如抽搐、昏迷、心跳停止、猝死、肝大、高氨血症等都很常见。
疾病突变位点:MCAD是由基因 ACADM编码,位于染色体 1p31,目前有 80 多种基因突变被发现,其中大部分是错义突变,位于第11外显子的985位碱基A突变为G(985A→G)为优势突变。
临床上疾病表现
MCADD常发于婴幼儿时期,患儿多出现呕吐、无精打采、低血糖等症状。MCADD患者也可能出现严重并发症,如癫痫发作、呼吸困难、肝脏问题、脑损伤、昏迷,以及意外猝死。

Copyright © 2016 中关村华康基因研究院培训 . All Rights Reserved京ICP备18042576号-2


